Post-Covid Neurological Symptoms
After contracting Covid-19 in August 2020, I developed a post-Covid syndrome with gradually increasing neurological manifestations. It took several months to turn into something that could be called a serious neurological condition without an evident cause.
I had a relatively mild form of Covid-19. I never had any chronic diseases that I was aware of, and I was a happy and energetic man in my late thirties. However, Covid changed that abruptly. Shortly after I was discharged from the hospital, I began to experience shortness of breath. It wasn’t happening all the time but was noticeable during physical loads and periodically at night when I was trying to get some sleep. During these episodes, I measured my oxygen saturation and it always was 99%. So, no reason to worry, right?
After the shortness of breath, I developed a form of a sore throat that was aggravated during the episodes of shortness of breath. I did not draw a link between the two at first, but consequent observations proved they were linked somehow. I was also fatigued and the fatigue lingered. I had an intellectually demanding job but could not work full time anymore. Five hours out of 8 were somewhat achievable but anything longer than that was problematic. I developed mild hypothermia as well. Instead of a normal temperature of 97.9 °F (36.6 °C) I always had 95.9 °F (35.5 °C). Finally, I also experienced intermittent episodes of slight tinnitus in the left ear.
I truly hoped these symptoms would be improved over time, but that did not happen. Moreover, in December 2020, four months after recovering from an initial infection, I began to experience what I can only describe as short and intermittent “halts” of consciousness. These episodes happened during work, especially when I was concentrating on a challenging task. The “halts” were like 1-2 seconds of fear that I was losing control over my body and was going to die. They happened every few minutes. If I relaxed a bit and moved away from the challenging task, the “halt” symptom weakened and disappeared.
In January 2021, I started to feel awkward. It was an unexplainable doom and gloom feeling of imminent death in the near future that was haunting me everywhere. I began to feel burning sensations in my right foot and right hand and my sore throat was becoming worse.
New Onset Panic Attacks
Meanwhile, I remained a big fan of walking and walked up to 8 miles (13 km) a day. After one such walk in February 2021, I experienced the first panic attack in my life. My heart rate went crazy and the shortness of breath reached its new maximum by turning into suffocation. I was thinking I was going to die and called the emergency.
I took every blood test I could imagine and visited every doctor I could reach. I went to a cardiologist, neurologist, endocrinologist, and phlebologist. Every test was negative. I was declared healthy except for a few seemingly unrelated things. I had a mild form of T2DM with A1C of 6.7 and insulin resistance with HOMA-IR of 7.8. My low-density lipoprotein levels were slightly elevated. An interesting observation is that I took blood tests right before the acute phase of Covid, and I had no T2DM back then. During a talk with an experienced endocrinologist, I was told that those relatively moderate levels of glucose in my blood could not wreak such havoc alone. It might be something else, but nobody knew what it was. A neurologist concluded that it was just severe depression and suggested taking some antidepressants, which I did without any positive results.
Meanwhile, my panic attacks intensified. The sore throat gradually transformed into dysphagia. The burning sensation in my right foot and hand took over the whole right side of my body. I started to lose the ability to speak fluently, and I started to have problems with my gait and street navigation.
At the height of a panic attack, I felt that my intrinsic biological processes were stopping and that one day it may have an ultimate ending. I started to develop a condition called intestinal ataxia. My right hand now had neurological edema. My peripheral neuropathy worsened significantly and became seriously painful. Everything went downhill very quickly, in a matter of weeks and days. At the beginning of April 2021, I faced an immediate risk of becoming an invalid and thought I might die.
The bitter part of the story is that nobody was going to help me. All the doctors I had met were helpless.
Strange Observations
I started to observe strange things while I was suffering from the condition. First, my symptoms slightly improved after consuming specific foods. For instance, it happened every time I ate a burger with real beef. The positive effect persisted for 2 or 3 days and then vanished. The same improvement I’ve experienced from consuming a glass of milk, but the effect was way shorter lasting just one day. Second, the condition was greatly improved on days when I consumed NSAIDs such as Aspirin or Ibuprofen for an unrelated reason. Third, I felt almost healthy on the rare days when I ate those specific foods and consumed NSAIDs together by pure coincidence. Unfortunately, I did not draw any specific conclusions back then. Such correlations just looked a bit weird to me as I never experienced anything like that in my whole prior life.
Discovering Poor Brain Glucose Metabolism
While the condition was gradually worsening, I measured my blood glucose levels several times a day. One day I decided to conduct an experiment to find out how blood glucose levels are affected by walking. I started by measuring my base glucose level in the morning which then was 126 mg/dL (7 mmol/L). Then, without eating anything, I decided to take an 8-mile (13 km) walk. To my amusement, the glucose level fell to 81 mg/DL (4.5 mmol/L) at the end of the route. Everything worked as expected, it seemed that I was not that insulin resistant after all.
But what about mental and intellectual activities? It would be a nice experiment to conduct too. The next morning, without eating anything, I went to my job with a glucometer and measured the base glucose level which was 117 mg/dL (6.5 mmol/L) at the time. Then I started to work taking the most challenging tasks. After 3 hours of intensive intellectual work, I started to experience the aforementioned “halts” of consciousness. Time to measure the glucose level. It was 115 mg/dL (6.4 mmol/L). Wait, what? How is that possible after all these intellectual activities? Yes, there is a process called gluconeogenesis that could raise the glucose level but still, I did not expect such a high value after such a massive cognitive load on an empty stomach. Clearly, something was going wrong with my brain as it had significant problems with glucose utilization. This was the crucial moment. A simple scientific experiment allowed me to see the light at the end of the tunnel. The cells of my nervous system were unable to consume the usual levels of glucose as they were insulin resistant, and I just proved that with my measurements.
Was I Deficient In Thiamine?
A quick web search of such a metabolic condition resulted in thiamine deficiency (vitamin B1) and beriberi as possible culprits. I knew about beriberi, but it never bugged my mind to link it with my own condition. I always thought it could be only caused by extreme malnutrition, which was not the case with me.
Going deeper on that route, I found the work of Hans Krebs, who described the process of cellular respiration and received the Nobel Prize for it in 1953. I was amused. Not only did it explain everything I had experienced, but now I had some actionable plan to try and improve my health.
If the usual levels of glucose cannot be consumed by the cells due to their insulin resistance, does it mean that by artificially raising glucose concentrations in the blood we can stop a metabolic panic attack? I conducted the experiment on several panic attacks of mine and received a positive answer. Yes, a metabolic panic attack can be stopped or at least significantly decreased by consuming 15 g of sugar. I immediately started to practice that to save my cells from further damage whenever a panic attack was mounting. That knowledge improved my condition a bit and gave me some time to find the appropriate therapeutic dose of thiamine.
Would thiamine help to reverse insulin resistance? I went to the pharmacy and bought Benfotiamine. It was hard to find, but luckily it was provisioned in two pharmacies in my hometown, so I did not have to wait for too long. Being in serious neurological suffering and pain, I immediately consumed 150 mg of Benfotiamine. Instinctively, I expected some kind of reaction, so I consumed the dose gradually. No reactions developed, but I immediately felt better in just 15 minutes after taking the pill. After that experience, I understood that I will be able to survive.
Given this experience and research, I wondered if the cellular damage would be reversed completely. It was an open question. Benfotiamine made me feel better, but I still experienced polyneuropathy and shortness of breath. The panic attacks went away completely, however.
Are Vitamins the Answer to Post-Covid Symptoms?
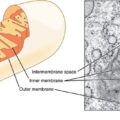
The new knowledge explained a lot of strange things that were happening with my body. Remember I talked about strange unexplained positive effects of consuming meat and milk? Now it became clear why: those foods contain nutrients including a rich set of vitamins and minerals. Vitamins help the mitochondria to process the substrates (glucose and lipids) to produce the energy (ATP) that powers up the cells.
It also explained the strange positive effects of consuming NSAIDs. As it turned out, this is relatively well known too. The mitochondrial dysfunction causes an auto-immune reaction of the body to its own metabolically subpar cells. This inflammation does more damage than good in that situation: the vessels’ endothelium gets damaged, leading to various blood flow problems including micro-clotting. This further aggravates the tissue hypoxia and makes the mitochondrial dysfunction even more severe in the affected areas, leading to even more inflammation. This is a self-fueling pathological process with a positive feedback loop.
An acquired mitochondrial dysfunction can be reversed to some degree by using supplementary vitamins, co-factors, minerals, and antioxidants. Thiamine takes one of the instrumental roles in this process as it catalyzes the reactions at the very start of the oxidative phosphorylation pathway, but thiamine alone may not do much unless a full spectrum of nutrients is supplied. Glucose is not the only fuel a mitochondrion can consume, and other pathways need attention as well.
Combining all these pieces and using the protocol for treating beriberi as the basis, I came up with an experimental therapy, which I first tested on myself. It consisted of B1, B3, B7, B2, multivitamin, magnesium, potassium, CoQ10, alpha-lipoic acid, resveratrol, L-carnitine, zinc, cuprum, and aspirin in certain forms and proportions. Going beyond medicals, it also included dietary corrections (ketogenic non-vegetarian diet, no tea, no coffee, and no alcohol) and mild physical activities (walking).
To my surprise and amusement, it gave the desired results. I was able to get rid of the panic attacks, hypertension, tachycardia, neurological edema, tinnitus, insulin resistance, dysphagia, cognitive impairment, fatigue, and neuropathy. It took some time and effort. One part of that was my own body that needed the time to adapt and heal, another part was numerous therapy refinements.
At the beginning of the therapy, I took frequent blood tests to ensure the right therapeutic direction. All my outstanding markers were gradually normalizing, proving that I was on the right track.
At the time, I was not aware of Drs. Lonsdale and Chandler Marrs’ work or Elliot Overton’s videos. You can imagine the level of my sheer astonishment when I compared my humble findings to theirs. Now, almost two years after the disease’s inception, I can call myself a healthy man, again.
We Need Your Help
More people than ever are reading Hormones Matter, a testament to the need for independent voices in health and medicine. We are not funded and accept limited advertising. Unlike many health sites, we don’t force you to purchase a subscription. We believe health information should be open to all. If you read Hormones Matter, and like it, please help support it. Contribute now.
Yes, I would like to support Hormones Matter.
Photo by Marjan Blan | @marjanblan on Unsplash.
This story was published originally on August 11, 2022.


You might be interested in