

Throughout the past few years, I have been prescribing thiamine more and more often for individuals with a range of different health conditions. I have witnessed major symptomatic improvement in some people who displayed none of the key risk factors for thiamine deficiency, and many times had been following clean, whole-food dietary regimes which contained levels of thiamine beyond what is suggested by the RDA. I began asking myself:
Why does thiamine, in sustained high doses, work so well for such a wide variety of diseases? Is it merely addressing a deficiency or is there something else going on here?
I have since come to the conclusion that one does not need to be deficient in the nutritional sense to benefit from this type of therapy; that high-dose thiamine is not simply working by correcting nutritional deficiency. Rather, thiamine is functioning as a metabolic stimulant to restore oxidative energy metabolism in cells that have been inhibited by factors unrelated to nutritional status.
Overwhelming toxicity and chronic oxidative stress have the capacity to inactivate thiamine-dependent enzymes involved in the generation of cellular energy, producing biochemical changes which are similar to clinical thiamine deficiency. This could basically be referred to as “functional” thiamine deficiency. In a functional deficiency, dietary thiamine intake is somewhat irrelevant, because the concentrations obtained via the diet are simply not sufficient to overcome enzymatic inactivation.
Instead, high concentrations of thiamine are often necessary to overcome the “metabolic block” and restore the deranged metabolism back to normal. Dr. Derrick Lonsdale has discussed this concept on many occasions and laid out the theory in his various writings. In this article, I will explain the rationale behind high-dose thiamine therapy as a tool for bypassing these metabolic blocks and examine how this can be a useful therapy for chronic health conditions.
Understanding Enzymes
To appreciate thiamine’s potential utility in mega-doses, we should first look at the very basic function of enzymes. Enzymes are a type of protein that the body uses as a catalyst to facilitate or “speed up” the rate of biochemical reactions.
Enzymes are responsible for driving the reactions involved in practically every known function of the human body, including building things up, breaking things down, modifying or changing molecules, and converting one molecule into another. Vitamins and minerals act as necessary cofactors or “helpers” for specific enzymes to work as they should. In the hypothetical diagram below, the enzyme responsible for converting substrate “A” into product “B” can only fulfill the task once it has bound its cofactor/coenzyme.
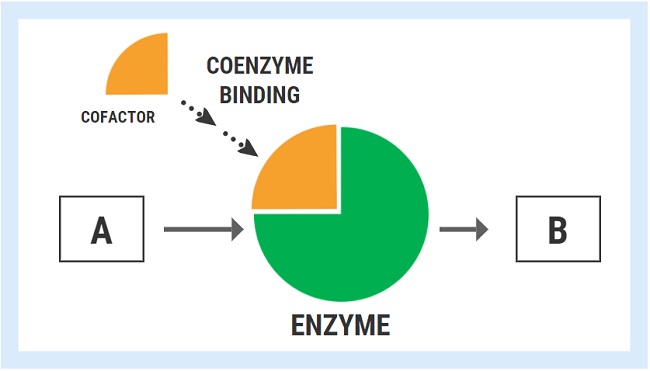
The ability of an enzyme to bind with its respective cofactor is referred to as the coenzyme affinity (km). A simple way to conceptualized this is to think of the enzyme like a magnet. Enzymes with high affinity for their coenzyme/cofactor exert a strong magnetic pull and can bind very readily with their coenzyme.
With high coenzyme affinity and more binding, the activity of the enzyme speeds up and the rate of reaction (A->B) increases. In contrast, enzymes with low coenzyme affinity exert a much weaker “magnetic” pull, meaning that they are less able to bind with the cofactor/coenzyme. Less cofactor binding means that the rate of reaction decreases.
Genetic Enzyme Defects and Nutrients
A variety of inherited, genetic conditions feature the production of defective enzymes with poor cofactor affinity. For these unfortunate individuals, the concentrations of nutrients found in food are simply not sufficient to overcome the genetically determined lack of affinity.
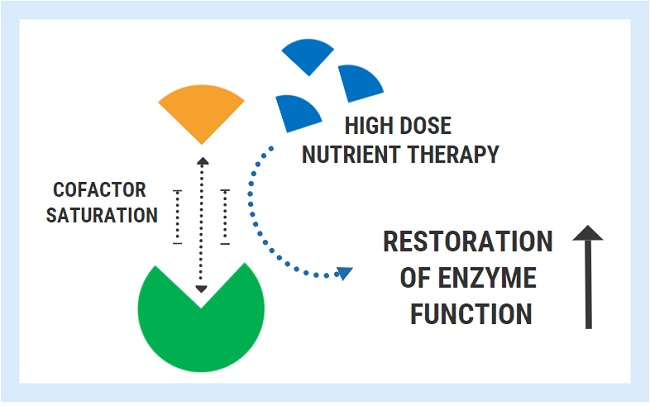
A successful strategy used for these conditions is the administration of pharmacologic/mega-doses of the nutrient cofactor. By saturating the cell, one can bypass the low affinity and restore enzyme function back to its normal state. Extremely high doses are often required to achieve this effect and this therapy must be maintained lifelong.
- Thiamine-responsive maple syrup urine disease: A genetic defect in the branched chain ketoacid dehydrogenase enzyme results in remarkably low affinity for its coenzyme TPP. Continued high doses are necessary restore the function of this enzyme complex.
- Thiamine responsive Leigh’s disease: Inherited mutation in the gene encoding Pyruvate Dehydrogenase, with a decreased affinity for its TPP cofactor. Treated with pharmacological doses of thiamine to stimulate defective enzyme activity.
- B12-responsive Methylmalonic acidaemia: Genetic defect encoding the methylmalonyl-CoA mutase enzyme, causing low affinity for adenosylcobalamin cofactor and a pathological accumulation of methylmalonic acid. This condition can be treated with megadoses of B12.
- Biotin-responsive holocarboxylase synthetase deficiency: Genetic mutation renders biotin-responsive carboxylase enzymes much less able to bind with biotin cofactor due to markedly decreased affinity. Supraphysiologic doses can restore normal enzyme function.
- B6-responsive homocysteinuria: A rare defect in the cystathionine-B-synthase enzyme reduces affinity for its coenzyme pyridoxal-5-phosphate. This leads to the toxic buildup of homocysteine. Mega-doses of vitamin B6 can return enzyme activity back to normal.
It is worth noting that these genuine genetic defects are extremely rare and are not applicable to the large majority of people. Nevertheless, similar principles can also be applied when an enzyme has been inactivated by other factors.
Prolonged Oxidative Stress, Inflammation, and Enzyme Activity
The activity of different enzymes is tightly regulated depending on metabolic requirements, energy intake, and numerous other conditions within the cell. In simplified terms, if cells need to break something down, build something up, slow a process down, speed a process up, the activity of the enzymes involved in those pathways will reflect that. Enzyme activation/inhibition is a necessary part of normal cell physiology. However, the activity of specific enzymes can also be affected by other factors including toxins. There are certain enzymes involved in energy metabolism which are particularly susceptible to inactivation by free radicals and oxidative damage. Short-term, this is most likely beneficial, but under conditions of chronic oxidative stress, such as that found in chronic disease, enzyme inactivation can become pathological.
A key enzyme involved in mitochondrial energy metabolism called alpha ketoglutarate dehydrogenase (KGDH). Several nutrients serve as cofactors for this enzyme complex, with thiamine taking center stage. KGDH is a rate-limiting step in in the TCA cycle, meaning that when this enzyme slows down, every other downstream step also slows down. Whilst a deficiency of any of the necessary cofactors will reduce the activity of this enzyme, it is also exquisitely sensitive to oxidative stress. KGDH appears to be more sensitive to disturbed homeostatic factors than other enzymes, playing the role of a metabolic redox sensor, capable of switching oxidative phosphorylation “on” or “off” depending on the cellular redox state and requirement for energy. Reactive oxygen species will selectively inactivate the KGDH complex and slow down oxidative energy metabolism. This inhibition is functionally beneficial for cells in the short-term as an attempt to avoid energy overload and oxidation. Not only is KGDH a target of oxidative inactivation, but it is also a significant generator of oxidative free radicals. Here, it plays a regulatory role which clearly serves essential functions in maintaining cell homeostasis.
Under long-term conditions of oxidative stress, chronic KGDH inhibition is thought to be a driving factor underlying many neurodegenerative diseases. In chronic fatigue syndrome, recent metabolomic analysis found that one of the few metabolites (out of 800+) elevated with statistical significance was alpha-ketoglutarate, which is perhaps also consistent with chronic KGDH inhibition. Several toxic and inflammatory factors have also been shown to inhibit KGDH. Immune cells in the brain called microglial are involved in neuroinflammation and can be activated by a variety of stressors including toxins, trauma, and infectious insult (think Lyme, or lipopolysaccharide coming from a leaky blood brain barrier). Microglia produce myeloperoxidase and downstream products including hypochlorous acid and mono‐N‐chloramine – all of which are powerful inhibitors of KGDH. Heavy metals including aluminum and arsenic, along with fungal mycotoxins inhibit thiamine-dependent enzymes including KGDH and pyruvate dehydrogenase (PDHC).
Activated microglia caused by inflammation in the brain generate excess amounts of nitric oxide and its free radical peroxynitrite, both of which further inactivate KGDH. Polyunsaturated fats lining neuronal membranes are prime targets for oxidative damage in the brain, yielding a toxic byproduct called hydroxynonenal (HNE). Once more, HNE was shown to inactivate both KGDH and PDHC, whereas other mitochondrial enzymes were unaffected.
Endogenous neurotoxins such as and isoquinolone derivatives (breakdown products of catecholamine neurotransmitters) have been associated with Parkinson’s disease, and also inactivate KGDH. These metabolites include oxidized derivatives of dopamine and norepinephrine. Other KDHC and PDHC inhibitors include the breakdown products of halogenated toxic chemicals such as Tetrafluoroethylene (TFEC).
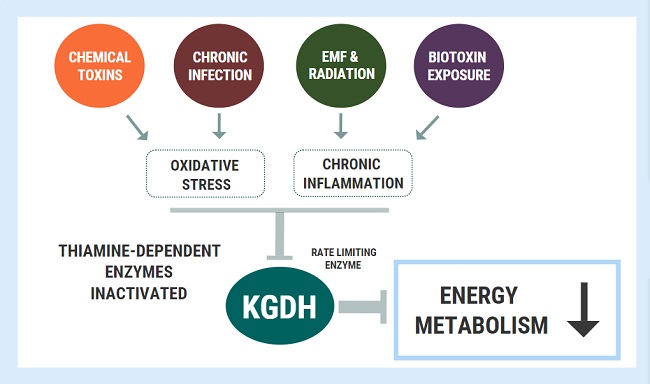
Oxidative stress and chronic inflammation are the hallmarks of chronic disease and both factors appear to inhibit/inactivate KGDH. As the rate-limiting step in oxidative phosphorylation, the chronic inhibition of this enzyme can spell devastating consequences for cellular energy turnover. A person could be obtaining a great amount of thiamine through their diet, but the underlying inhibition of these enzymes will produce the exact same outcomes as a dietary deficiency. In other words, these changes will induce a functional deficiency.
Mega-Dose Thiamine to the Rescue
When enzyme inhibition becomes pathological, we can apply similar principles as outlined above with nutrient-responsive genetic conditions. We can use high doses to bypass or overcome the metabolic blocks caused by enzyme inhibition. This concept was wonderfully illustrated in a study titled: Thiamine preserves mitochondrial function in a rat model of traumatic brain injury, preventing inactivation of the 2-oxoglutarate dehydrogenase complex.
For this study, researchers investigated the effects of traumatic brain energy (TBI) on energy metabolism, using several groups of rats who were not deficient in thiamine. They showed that the oxidative stress associated with TBI inactivated the KGDH enzyme, causing great reductions in energy synthesis, which was coupled with brain damage. Administering massive doses of thiamine to the rats before TBI was able to completely protect the KGDH enzyme. The thiamine-treated group maintained normal activity of KGDH, mitochondrial respiration, and ATP despite being exposed to the injury. Furthermore, the restoration/protection of KGDH might have also conferred some degree of cytoprotection by combating inflammation, which was demonstrated by reduced inflammatory gene expression at three days post-TBI.
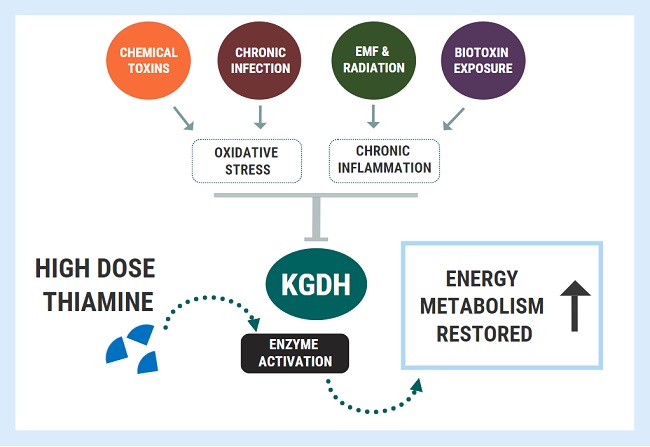
What this study demonstrated was that very high doses of the cofactor could provide protection against an insult which was not related to deficiency. In fact, similar results have been shown in several other studies:
- Thiamine administration protected neurons against inflammation-induced impairments in neurogenesis caused by exposure to radiation, both in vitro and in vivo. Thiamine treatment also significantly increased lifespan. Attenuation of these inflammatory effects are thought to be due to increased stimulation of KGDH activity.
- A more recent study also looked at traumatic brain injury (TBI) with a focus on glutamate neuroexcitoxicity. They showed that excess nitric oxide and peroxynitrite found in neuroinflammation led to the inactivation of KGDH. KGDH inhibition reduced glutamate uptake into the Kreb’s cycle, producing glutamate excitotoxicity and neuronal cell death. Once again, extra levels of thiamine reversed this issue by stimulating KGDH, increasing glutamate clearance and protecting the cells against injury. The authors concluded:
Thus, the impairment of OGDHC [KGDH] plays a key role in the glutamate mediated neurotoxicity in neurons during TBI; pharmacological activation of OGDHC may thus be of neuroprotective potential.
Interesting choice of words, huh? They are basically telling us that the pharmacological use of thiamine might be helpful in conditions where KGDH is inactivated, and enzymatic stimulation can be protective against glutamate neuroexcitoxicity. For the reader’s reference, here are a quick list of conditions which are thought to involve neuroexcitoxicity as part of the disease-process:
- ME/Chronic Fatigue Syndrome
- Amyotrophic lateral sclerosis, Huntington’s and Alzheimer’s diseases
- Depression, Autism and Schizophrenia
- Parkinson’s and Multiple Sclerosis
- Electromagnetic hypersensitivity syndrome
- Multiple chemical sensitivity
- Lyme disease
Spinal cord injury leads to significant neuroinflammation similar to that found in TBI, with excess nitric oxide production and deficits in brain glutathione levels (an intracellular antioxidant). In one study: thiamine in high doses ameliorated excess nitric oxide levels and maintained brain levels of glutathione. The authors hypothesized that this was related to changes in precursor amino acid availability. However, this is likely also related to the stimulation of transketolase (TKT) activity (a thiamine-dependent enzyme involved in replenishing reduced glutathione). Under conditions of oxidative burden and increased requirement for glutathione recycling, there is a need for increased TKT activity and thiamine.
High doses of thiamine will stimulate the transketolase enzyme to maintain glutathione levels. This was shown in a different study using metabolomic analysis in cardiac ischemia, which found increased levels of ribulose-5-phosphate suggestive of increase TKT activity. Indeed, both thiamine and benfotiamine were found to increase the genetic expression and activity of the transketolase enzyme to counteractive oxidative damage and cell injury in diabetic vascular endothelial dysfunction. High doses of thiamine can also restore activity of the pyruvate dehydrogenase enzyme complex in the face of inactivation. Cardiac arrest was shown to markedly depress PDHC activity through inactivation.
In rats, high-dose thiamine post-cardiac arrest restored pyruvate dehydrogenase activity in brain, mitochondrial respiration, improved neurological function, reduced brain injury, and improved survival at 10 days. The quantity of the enzyme did not change, showing that thiamine worked by stimulating PDHC activity at high doses, thereby preventing injury-induced inactivation of this enzyme complex.
Pre-treatment with thiamine pyrophosphate protected against cardiac ischemia by maintaining mitochondrial function, ATP concentrations, and inhibiting mitochondrial fission.
Furthermore, copper toxicity was shown to inactivate the PDHC , produce mitochondrial dysfunction and neurological damage in rats. High doses of thiamine protected against the inhibition of Pyruvate dehydrogenase, markedly extended life span and protected against neuronal death.
The Use of Mega-Dose Thiamine in Clinical Practice
The late Italian neurologist A. Constantini published several case studies on the use of mega doses of thiamine for different conditions and saw impressive results. In one of the case reports on fibromyalgia, two patients saw an abrupt and immediate improvement only when they reached 1,800mg per day. At lower doses, improvements were negligible. High dose thiamine produced appreciable improvements in fatigue in 15 MS patients. Likewise, high doses were shown to produce remarkable and rapid improvement in the neurological condition essential tremor. Severe chronic fatigue in IBD patients with normal thiamine lab tests was reversed in most patients with megadoses.
Thiamine injections completely reversed gait abnormalities and motor failure in two patients with Freidrick’s Ataxia. Importantly, Constantini and colleagues concluded:
From this clinical observation, it is reasonable to infer that a thiamine deficiency due to enzymatic abnormalities could cause a selective neuronal damage in the centers that are typically affected by this disease.
Furthermore, in a case report of two patients, dystonia was reverse with thiamine administration. I have also seen this occur in several children with autism and/or neurodevelopmental abnormalities. Another case report detailed high-dose thiamine injection in patients with Parkinson’s disease, all of which had “normal” plasma thiamine levels, meaning that they were not classically diagnosed as having deficiency. The patients experienced between 30 and 77% improvement in motor coordination. We have seen from the research above that the neurotoxic metabolites which are thought to drive Parkinson’s also have the strong capacity to inhibit thiamine-dependent enzymes. It is therefore no wonder why thiamine can have such as tremendous impact on this condition.
Constantini and colleagues completed a larger study with 50 patients two years later, and found that 100mg thiamine injection twice per week produced massive improvement in both motor and non-motor symptoms, with some patients experiencing complete clinical remission.
Are these results simply addressing a deficiency or is something else going on here?
The daily recommended dietary intake of thiamine is merely 1 – 1.5 mg per day. Surely, if the benefits were simply due to nutritional repletion then we would see benefits at similar levels, or even 10x that amount? Except we do not. Rather, most people are required to consume 100 to 1000 times the daily recommended intake to see restoration of metabolism and symptomatic improvement. This is what I see in clinical practice on a frequent basis, and this is also what has been demonstrated in the case literature.
Beyond Treating A Deficit
The sheer amount of the nutrient necessary for clinical improvement is not consistent with simply addressing a deficit. Nutritional repletion is by no means an adequate explanation for this magnitude of effect. It IS consistent with stimulating enzyme activity to overcome inactivation, however.
Constantini hit the nail on the head with one quote from another paper:
We may suppose that symptoms decrease when the energetic metabolism and other thiamine-dependent processes return to physiologic levels. Our aim was not to correct a systemic deficit of thiamine, but rather to increase the activity of enzymes involved in cell production energy in selective brain regions.
Indeed, Constantini understood that thiamine could be used as metabolic enhancement to stimulate the enzymes involved in energy metabolism which had otherwise been inhibited by other factors. This is where we are dealing with a “functional deficiency” which can only be addressed by supraphysiologic concentrations to saturate the cell for improved bioenergetics. As I said mentioned previously, Dr. Derrick Lonsdale has highlighted on many occasions how thiamine’s effective is due to its pharmacological action, rather than nutritional repletion.
Rather than remaining hyper-focused on correcting a deficit, we should be using this molecule to improve bioenergetics regardless of nutrient status. This means that someone does not necessarily need to be nutritionally deficient to benefit from thiamine supplementation at high doses.
The Non-Enzyme Functions of Thiamine
It is worth noting here that there are a few other variables which I have not discussed thus far. Outside of the context of genuine inherited genetic defects, there are numerous polymorphisms in genes related to thiamine transport and metabolism. These polymorphisms can influence enzyme activity, albeit to a lesser extent, and can predispose one to developing a deficiency. Nonetheless, this does not alter the fundamental principles laid out in this article. It is also important to understand that the clinical improvements demonstrated are not just due to thiamine’s role as a cofactor to drive biochemical reactions to their completion. Rather, this nutrient exerts numerous non-coenzyme functions including allosteric regulation of other enzymes in energy metabolism, direct anti-oxidant and anti-inflammatory actions. It has been shown to influence the transcription of genes involved in modulating and dampening inflammation and oxidative stress upstream. Thiamine and benfotiamine supplements exhibit “anti-stress” properties in the brain, protecting against stress induced suppression of hippocampal neurogenesis. These effects stem from anti-oxidant, rather than coenzyme roles. A review of thiamine’s non-enzyme actions can be found here.
Thiamine as a Front Line Therapy
At this point, I hope that the reader can appreciate some of the potentially beneficial applications of thiamine therapy in high doses. Since this nutrient exhibits extremely low toxicity, is relatively cheap and easy to access, I believe that it should be considered as a front-line therapy, in conjunction with other interventions, for disorders involving mitochondrial dysfunction and chronic oxidative stress. This especially applies to neurological diseases. Whilst many people do not require pharmacological doses, there are many who DO benefit from this. I have seen it on many occasions, and I am sure that I will continue to do so in the future. As it currently stands, the therapeutic potential of this nutrient is untapped.
Disclaimer
I should note, before beginning any type of treatment, consult your physician. The work above represents the current state of research and observations from my own clinical practice. It should not constitute medical advice. Please be aware that although this vitamin is non-toxic and one cannot overdose, some individuals with longstanding health issues exhibit negative reactions upon taking even small doses of thiamine. These reactions are often associated changes in electrolyte homeostasis, other nutrient deficiencies and/or can be associated with the formulation of thiamine administered. There are a number of articles on these reactions on this site under the search terms, paradoxical reaction, refeeding syndrome, and more recently, calcium management and heart function.
We Need Your Help
More people than ever are reading Hormones Matter, a testament to the need for independent voices in health and medicine. We are not funded and accept limited advertising. Unlike many health sites, we don’t force you to purchase a subscription. We believe health information should be open to all. If you read Hormones Matter, like it, please help support it. Contribute now.
Yes, I would like to support Hormones Matter.
Photo by Jack Carter on Unsplash.
This article was published originally on EONutrition on November 17, 2020 and edited and republished here with permission.












Hi Elliot,
Thanks for all the work you’ve done it seems like you’ve helped so many people. I’m trying to fix my severe sleep maintenance insomnia / sleep apnea. The apnea is diagnosed as ‘mild’ only an AHI of 6 but the effects are devastating on me, I need 10+ hrs sleep every night and still need multiple naps a day just to function, it’s truly horrific and that’s when I sleep through the night.. often I also wake up a couple hours before I’m supposed to and am unable to fall back to sleep also.
Ive seen some anecdotal evidence of thiamine working on sleep apnea and I’ve seen one mention from you about it but not much more information that that unfortunately. I just want to go back to sleeping 8 hrs and feeling incredible again like I did only a few years ago. Apart from my sleep issues and the apnea, I also have a very bad DHEA and pregnenolone deficiency revealed by blood tests (I’m sure the stress of the insomnia and apnea caused this) I’m now on HRT for this and it fixed up some of the weird issues I was dealing with, like stress intolerance and light sensitivity and wired feeling at night. The only other symptoms I suffer from is reoccurring constipation every month or two in which I get bad pains in my right side and have to take suppositories to clear me out and cold hands and feet.
Can you point me in the right direction in terms of how I can go about dosing and any cofactors I may need for my specific case, you don’t seem to go over anything that will fix my main issues (sleep) in your thiamine protocol.
Thanks again,
Charlie
I have UARS, thiamine removed my brainfog and allowed me to be able to think again. Do it.
I have a ALS and I am taking alpha ketoglutarate per the Diana protocol. I am wondering how the alpha ketoglutarate dehydrogenase you talk about figures into this. Am I harming myself by taking the AKG? I am also working my way up with the TTFD to 600 mg per day per Dr. lonsdale‘s recommendation.
Good morning, I read ALL the posts on this blog, it’s just magic! I have also watched ALL of Elliot’s videos. I wonder if there is the possibility of creating a guide for doctors worldwide who know the benefits of thiamine and work with patients on an individual level. I’ve been taking it for 5 months and it changed my life, I’ve lived tired all my life, fortunately I haven’t had any diseases despite my low energy level. But after 5 months taking it I have lost energy again and I do not understand what has happened, I do not want to live like this knowing what it is like to live normally with energy. I take TTFD (150mg) and benfotiamine (250mg) and I accompany it with complex B, magnesium, potassium, biotin, B2, carnitine and I usually take betaine beforehand, since I have very low stomach acid. Is there any explanation? I have found a laboratory that does the transketolase test, would it be worth doing? Or should I seek genetic counselling? I will continue to take it daily. Thank you.
update! I took acetyl carnitine again and in a matter of minutes I recovered the energy that I had lost days ago. I understand that I need both thiamine and carnitine for a “normal” level of energy. Where will be the problem?
Elliot, thank you for this excellent detailed and well researched article. Dr. Lonsdale thank you for your years of dedication to thiamine. Having a western allopathic diagnosis of MS and having treated chronic Lyme in the past with my naturopath, plus now having extremely high B12, I suspect that I have several functional B deficiencies going on. I have been taking 50 mg thiamine hydrochloride and 150 mg benfotiamine and 5 mg of lithium orotate. I’ve only started recently, but I have more energy than I think I have ever had in my life and I am 45. I am encouraged beyond belief. The more I read here and in the FB group the more things makes sense biologically. I plan to introduce allithiamine eventually as well, but since I am sensitive to methyl donars I am tweaking my other B vitamins/cofactors first before moving up/adding more of any type of thiamine. I am beyond grateful for you two and Dr. Chandler. I have shared this information with my two best friends, sister and my mom all of who have some kind of mitochondrial energy health related issues. I have also shared this with my naturopath and will continue to sing thiamine’s praise and point everyone here for the details. Thank you, thank you.
Dr. Lonsdale, I am reading these articles and finding them of great interest and help. I’m sharing them with doctors and laymen alike. Thank you. — brocktoon
The opinion of most people is that modern medicine is a glorious peon to the success of science. I would certainly agree that surgery has indeed come a long way. However, removal of sick organs from the body constitutes an admission of medical failure. Only a small number of interested people are reading these posts and it is going to take a long time for these words to be recognized as “the truth”. The idea of nutrients for treatment is hardly new because it was advocated by Hippocrates as long ago as 400 BCE. I would urge readers of these posts to discuss them with their friends and try to spread the word. Unfortunately very few physicians are aware of the magic represented here by Overton and others.
Any chance that you, Elliot, or Marrs has a recommendation for where to learn more about these enzymes and metabolic pathways along with all the necessary cofactors. I’m taking some vitamin/nutrient courses but I want to learn how it all works. Is this molecular biology that Elliot is talking about or biochemistry (or both or neither) and do I need to go back to college for this or is there a really good online course I can take or education I can obtain. I’ll spend the money if necessary but I think we’re in agreement that the time spent on microbiome in a lot of these courses is time and money somewhat wasted, and that it’s the energy models we need to focus on. I’m just a certified health coach heavily steeped in nutrition and corrective exercise and I get most of my clients really good results but after implementing thiamine and other vitamins and minerals into my protocols based on all the information I’ve learned from you three, I agree that this should be a big focus in the health space and I’m in100%. I just don’t have the knowledge that Elliot or you guys do and need to know the best and most efficient place to obtain it so I can continue on this journey and help others. I have huge dreams, scary dreams that if I can achieve, will change the world. Any advice on how to fast track obtaining this knowledge would be greatly appreciated and yes I have read and own your book. I just need to understand all the other pieces.
Thank you in advance.
This is such an excellent article!!! Thank you so much–I am a layman and have to spend more time wading through this, but wanted to say I am really encouraged that there are doctors out there working with thiamine. Thank you for writing this!