

Individuals who have been chronically ill often show varying degrees of thiamine insufficiency ranging from functional deficiencies where, due to the energetic demands of an illness, diminished capacity of transporters and/or enzymes, and/or the high degree of oxidative stress, the need for thiamine outstrips the supply available via diet alone, to quite severe and frequently longstanding frank thiamine deficiency disease processes like beriberi or Wernicke’s encephalopathy. Depending upon the origins and chronicity of the deficiency, thiamine replenishment outside of hospitalization can be challenging and yet this is where it must take place, as physicians are reticent to consider or treat thiamine deficiency in office or hospital. Thiamine deficiency is neither recognized in the population nor accorded the resources required to resolve and as a result, patients are left to navigate the illnesses brought on by thiamine deficiency independently.
For some, thiamine supplementation is a simple process that nets noticeable improvements in symptomology almost immediately. For others, however, the process is grueling and marked by a period of time where symptoms become noticeably worse before improvement is seen. Replenishment of thiamine in longstanding deficiency, particularly when the heart is affected, is prone to a variety of ‘refeeding syndrome’ – type responses. Conventionally, refeeding syndromes involve disturbances of potassium, magnesium, and phosphate and develop when nutrients are re-introduced after a period of absence-based malnutrition, e.g. some form of starvation (see Figure 1.).
Figure 1. Refeeding syndrome.
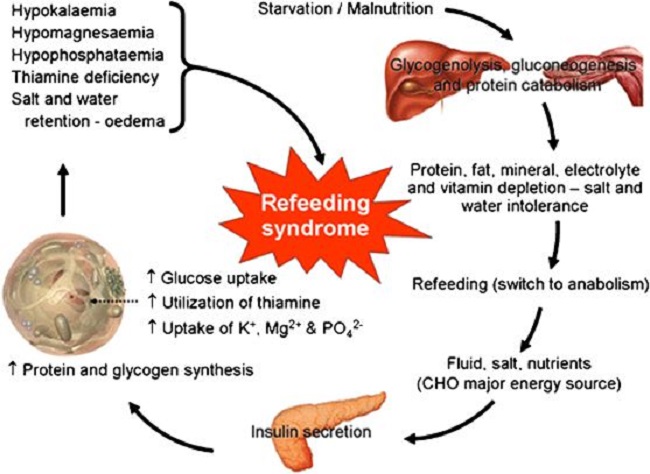
Symptomatically, these disturbances clearly affect heart function with troubling changes in rate, rhythm, and pressure. I suggest that the responses we see with more modern examples of chronic thiamine deficiency are generally caused by a higher calorie type of malnutrition; one where sufficient calories may be ingested absent sufficient nutrients. This is process is likely to be metabolically different than that of starvation or malnutrition. This means that while electrolyte disturbances often coincide, there may be other factors involved as well; factors that have yet to be fully understood.
Although the symptoms of refeeding noted in the high-calorie types of malnutrition may cause similar disturbances to heart function as the starvation-originated refeeding syndromes, given the distinct metabolic origins of these processes, different responses may be necessary. In some cases, the refeeding-like syndrome may be related to the formulation of thiamine used to treat the deficiency. There are several reasons for this, and our good friend Elliot Overton has begun to elucidate some of the chemistry involved. Across formulations, however, for a subset of patients thiamine repletion distinctly affects heart function even at micro-doses. Given that these patients are notably deficient in thiamine, that thiamine deficiency is absolutely requisite for mitochondrial function, for heart function, and for life itself, and its absence is directly associated with heart failure, how does one replenish thiamine when even the smallest dose sets off a cascade of troubling reactions?
With conventional treatment of wet beriberi, thiamine deficiency that affects the function of the heart, high doses of IV thiamine are provided for several days to a week in hospital, followed by high doses of oral thiamine for an extended period of time. The literature is rife with examples of success stories following this type of protocol. Absent from these case reports are instances of negative cardiac reactions to the high-dose thiamine. This suggests either these types of reactions do not occur by some function of the higher dosage, that they do occur but are mitigated by close consideration of electrolyte balance and/or other variables, or that they do occur but are simply not reported. Whatever the reasons, this would be useful information to practitioners and patients alike.
Assuming that these types of reactions occur, it is not clear why or by what mechanisms, beyond, of course, the electrolyte disturbances noted in refeeding of formerly starved individuals. This is important because while traditional interpretations of thiamine deficiency consider it rare and specific to a particular subset of patients for whom general nutrition and nutrient absorption is problematic (alcoholics, gastric bypass patients, parenteral feeding, anorexics, poverty-based malnutrition/starvation, pregnancy, with and without hyperemesis, and critical illness), it is not clear how those who develop beriberi in the presence of sufficient caloric intake, but with longstanding illness, differ metabolically from the traditional patient reported in the literature. And really, if we are honest with ourselves, we do not know much about the processes leading up to what are considered the more emergent cases of thiamine deficiency or even about the cases of heart failure where thiamine deficiency was likely a contributing factor but never considered. We have these pat, fully enshrined ideas about the ‘what and why’ of thiamine deficiency but rarely consider if or whether we know what we think we know.
With all of that in mind, I have been looking into potential mechanisms involved in the remodeling of heart tissue and function relative to longstanding thiamine insufficiency and the problems associated with thiamine treatment in these patients. I have observed that for some individuals, including those who have tested positive for thiamine deficiency and who clearly need thiamine, the addition of even the minutest amount of thiamine sets off a cascade of reactions that affect the pressure, rate, and rhythm of the heart. In these individuals, who may or may not have had previous, or at least previously recognized symptoms associated with cardiac function, the irregularities pose a difficult challenge for recovery; one for which the current literature has few direct answers.
I suspect these reactions involve an inability to smoothly switch from a previously hypoxic state to a marginally more normoxic one. I suspect also that the problem involves disturbed calcium (Ca2+) management adapted to the hypoxic environment that is then forced to re-equilibrate rapidly once respiration and oxidation kick on. Perhaps the response is akin to an ischemia/reperfusion process but one where longstanding molecular adaptations are involved versus an acute constriction of blood flow and oxygen. Regardless of the difference in time and scale, both the cause and the consequence involve altered Ca2+ flux, which is arrhythmogenic. If this is the case, it still begs the question about how we proceed with thiamine repletion, and perhaps also, how do we identify those most likely to respond negatively to thiamine? I do not have the answers to those questions yet, but perhaps if we unpack the relationship between mitochondrial function and calcium regulation we might find clues.
Mitochondrial Competence and Calcium Regulation
Calcium regulation depends upon functioning mitochondria and a steady stream of ATP. Mitochondrial functioning, in turn, depends upon the appropriate concentrations of Ca2+. The relationship between the two is reciprocal and dynamic. Failure in either leads to failure in both.
Mitochondrial nutrient deficiencies lead to reduced energy metabolism and utilization and ultimately molecular hypoxia. Since the heart requires an enormous amount of energy to maintain constant contractility, upwards of 15 times its weight in ATP – or about 6kg, every single day, mitochondria play a prominent role in heart function.
In the cases I see, presumed issues with Ca2+ mismanagement likely correspond to longstanding deficits in mitochondrial energetics evolving from some degree of metabolic inflexibility of the cardiomyocyte specifically, but likely, systemically. This metabolic inflexibility is both a consequence and a cause of insufficient thiamine, and likely other nutrients, that ultimately depress mitochondrial function and the capacity to produce ATP. The reduction in ATP then evokes a cascade of adaptive reactions, among them is a switch in fuel preference in the heart itself, from fatty acid oxidation to an increased reliance on glucose and anaerobic glycolysis; patterns consistent with systemic metabolic disruption. Additionally, the breakdown of amino acids is disrupted, leading to an accumulation of branched-chain amino acids, which further imperils energy production but also protein synthesis cascades. Finally, the decreased oxidative capacity that diminishes ATP output then continuously influences and is influenced by, Ca2+ dynamics creating a cycle of dysfunction. Arguably, it is these changes in mitochondrial metabolism that force alterations to cell and tissue morphology leading to the grossly observable pathology in later-stage heart failure.
Calcium Dynamics, Mitochondria, and the Heart: A Deeper Dive
Calcium dynamics in the heart are directly tied to the mitochondria at a number of junctions. Broadly, at physiological concentrations, Ca2+ activates oxidative phosphorylation (OXPHOS), e.g. ATP production. At supraphysiological concentrations, however, as we might see locally in the cardiomyocyte during the progression of heart disease to heart failure, Ca2+ inhibits OXPHOS. The inhibition of OXPHOS would presumably drive the shift in fuel source ultimately associated with heart failure. It would also drive calcium uptake, both by the mitochondria and sarcoplasmic reticulum (SR), altering cardiomyocyte contractility patterns. As mentioned previously, Ca2+ management by the mitochondria and SR are profoundly energy/ATP intensive processes.
Decrements in ATP availability, then lead to the poor uptake of Ca2+ by the mitochondria and SR, which then results in excess intracellular Ca2+ and impaired relaxation of the cell. This leads to high oxidative stress and increased reactive oxygen species (ROS) output; a pattern likely seen systemically. The high oxidative stress load caused by the overload of Ca2+ eventually stimulates mitochondrial permeability transition pore (mPTP) opening and mitochondrial swelling, resulting in mitochondrial injury, apoptosis, cardiac remodeling, and ultimately, the development of heart failure. Interestingly, a study in rodents found that with marginal thiamine insufficiency, the ensuing accumulation of pyruvate opened the mPTP but in a low-conductance state that apparently does not lead to cell death. The authors postulate that this may be a protective response that tempers the excess pyruvate and conditions the heart to reduced oxygenation.
Among the mechanisms by which Ca2+ regulates OXPHOS, all are nutrient-dependent; nutrients that are likely insufficient in metabolic disease and the progression to heart failure (see Figure 2.).
Figure 2. Mitochondrial nutrients.
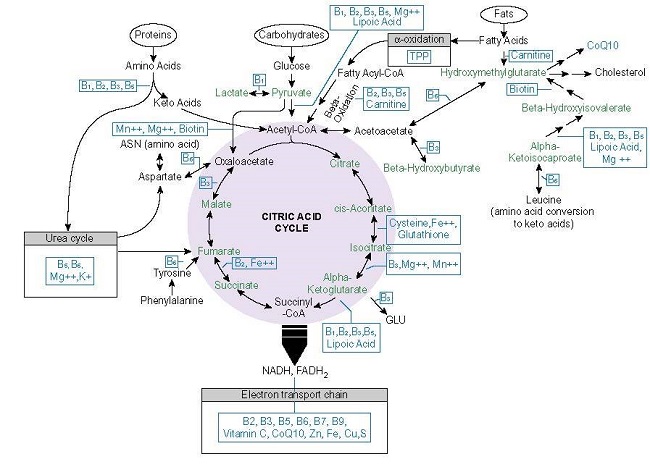
Moreover, each of these enzymes is activated appropriately only in the presence of the requisite nutrients and at low levels of Ca2+. At a higher concentration of Ca2+ and/or in the absence of the other nutrient cofactors, Ca2+ inhibits these enzymes, effectively shunting ATP production.
- Calcium activates the pyruvate dehydrogenase complex (PDC) both directly by binding to pyruvate dehydrogenase, or indirectly by upregulating inactivation processes and binding to pyruvate phosphatases. The PDC is exquisitely sensitive to thiamine and magnesium.
- Calcium stimulates the alpha-ketoglutaric dehydrogenase enzyme complex (KGDH), a rate-limiting enzyme within the Krebs cycle, but only at low concentrations and in the presence of magnesium and thiamine. At high concentrations and in the absence of sufficient Mg2+ or thiamine, Ca2+ inactivates KGDH leading to impaired energy metabolism and poor calcium management in the mitochondria.
- Calcium upregulates mitochondrial complex I, III, IV, and the Vmax of complex V, leading to KGDH inhibition when appropriately operational, and once again, likely only in the presence of the requisite nutrient cofactors (see Figure 2).
- Each of the processes above is energy dependent, necessitating sufficient ATP, but also, use the energy released by these reactions to reduce the electron the carriers nicotinamide adenine dinucleotide (NAD+) to NADH and Flavin adenine dinucleotide (FAD+) to FADH2. Both NAD and FAD require niacin and riboflavin, respectively. Calcium activates FAD-glycerol phosphate dehydrogenase and NAD-isocitrate dehydrogenase, but again, only at low concentrations and in presence of sufficient ATP and magnesium.
In sum, the relationship between the mitochondria and Ca2+ regulation is largely dependent upon the presence of critical micronutrient cofactors. Insufficiencies imperil ATP production and Ca2+ management, which then leads to a cycle of dysfunction likely at play in heart failure.
Calcium Capture and Release by the Sarcoplasmic Reticulum
If we move out of the mitochondria into the cytoplasm, we see similar sequences of events play out in the SR. The SR within the cardiomyocyte bears the brunt of responsibility for Ca2+ management in normally functioning hearts. Indeed, some research suggests that it is only when things go awry that the mitochondria step in to become the primary Ca2+ sinks, sequestering the excess. Otherwise, mitochondrial Ca2+ stores reflect those of the cytosol due to the constant influx and efflux of the cation via a number of transport channels, uniporters, and exchangers.
Here again, Mg2+ is involved. Magnesium, it appears, competes with Ca2+ at the ryanodine receptor, a critical Ca2+ receptor in the SR, to inhibit the release of intracellular Ca2+, and essentially temper Ca2+ activation/release. As with the mitochondria, SR activation is possible only at low concentrations of Ca2+ and when Mg+ and ATP are complexed together. High concentrations of Ca2+ inactivate the receptor, inhibiting the further release of Ca2+ into the cell. Similarly, high concentrations of Mg2+ also inhibit the receptor. All of this has to be exquisitely timed and balanced. Too much or too little, too soon or too late induces asynchrony in the contractility of the heart muscle and is one of the variables in the progression to heart failure.
Of note, caffeine increases the sensitivity of the ryanodine receptor by ~20-50 times. This means that less Ca2+ is required to open the receptor and release Ca2+ stores, potentially leading to high intracellular Ca2+ levels. Coffee and tea, the two more popular carriers of caffeine, are also high in thiaminase, a thiamine breakdown enzyme. Given how many of us are addicted to coffee and the caffeine it holds, this is problematic for both thiamine maintenance and heart function.
Problems with Thiamine Repletion
With all of this, I still have no idea what is causing the negative reaction to thiamine repletion in some patients. I do have a few ideas though, but none are fully worked out just yet. My gut tells me it is a matter of too much too soon and an inability to smoothly switch from the previously hypoxic state that relied on anaerobic glycolysis for ATP to a more normoxic state and an upregulation of OXPHOS. To that end, I suspect that the problem rests with three variables, the lack of substrate fuels for the newly upregulated OXPHOS (glucose and fatty acids), a lack of additional micronutrients (riboflavin, niacin, and magnesium, but possibly also Coq10) and an inability to manage the increase in carbon dioxide resulting from the increased oxygen utilization. All of this, of course, would impact Ca2+ homeostasis. I suspect the key roadblock in the process is alpha-ketoglutaric dehydrogenase, both as a rate-limiting complex in ATP production, ROS regulation, and Ca2+ regulation, but also, I suspect it is somehow tied to the regulation of the carbonic anhydrase enzymes that manage blood gas exchange. How though, I am not sure.
First possibility: Thiamine kick starts previously quiescent enzymes increasing ATP production via OXPHOS and Ca2+ induced excitation begins. Rather than appropriately sequester excess Ca2+, the longstanding reduction of Ca2+ in the cell is suddenly upregulated by SR receptor open time. That then feedbacks and shuts off the alpha-ketoglutaric dehydrogenase complex, which would again reduce ATP, while simultaneously increasing ROS. Alpha keto glutarate is a primary anti-oxidant next to glutathione. Elevated ROS and mitochondrial Ca2+ underpin several variables in heart failure including hypertrophy, fibrosis, and contractile dysfunction. If thiamine is given with insufficient magnesium and the other nutrients required for proper mitochondrial functioning (Figure 2.), alpha-ketoglutaric dehydrogenase would become inhibited or asynchronous to the relative increase in thiamine and Ca2+.
Second possibility: the increased thiamine fails to initiate OXPHOS sufficiently and continues to feed the anaerobic cycle shunting more pyruvate to lactate dehydrogenase with an ensuing buildup of lactate and a yet insufficient amount of ATP. With longstanding thiamine deficiency and metabolic dysfunction, the fuel preference of the heart adapts and switches from a primarily fatty acid-derived form of oxidation (60-70% fatty acid preference over glucose) to one that is entirely dependent upon large amounts of glucose via anaerobic fermentation. The shift back would be difficult, especially if thiamine is given quickly and in large amounts, or possibly any amount, absent the other nutrient co-factors. While thiamine is critical in the first steps of fatty acid metabolism, in alpha oxidation, to get through beta-oxidation, one needs carnitine, and vitamins, B2, B3, and B5. Absent those co-factors, and importantly, absent the substrate fatty acids to metabolize, we might yet get held up. Conceivably, this might also happen absent sufficient glucose to sustain the long-entrenched baseline production of ATP via anaerobic metabolism while OXPHOS is upregulating. While glucose is typically contraindicated in the thiamine repletion, as it is often a causative factor in the shifted metabolism in the first place, it is possible that in some instances it may be warranted. If the heart’s preferred substrate is not available then no matter how much we upregulate the OXPHOS pathway, energy production would falter.
Third possibility: Here, I have not yet fully put the pieces together, but briefly, it is likely the longstanding thiamine deficiency changes the regulation of blood gases and that this shift is problematic when oxygen status changes. We know that thiamine deficiency stabilizes the hypoxia-inducible factor (HIF) proteins. HIF proteins are the key regulators of the hypoxia cascades. That means, the absence of thiamine causes a sort of molecular hypoxia, an inability to utilize oxygen for the production of energy and to traffic it appropriately in the hemoglobin >oxyhemoglobin. In the case of longstanding thiamine insufficiency or deficiency, we have all of these underlying adaptations that have come into play to compensate, including those involved in the management and usage of oxygen. Since thiamine modulates this process and has been noted to be a carbonic anhydrase inhibitor both deficiency and excess would be problematic. I suspect that when we shift one side of the equation e.g. add thiamine back to the process, effectively upregulating the use of O2 and the production of ATP, the compensatory reactions of the other side, the re-regulation of Ca2+ and the carbonic anhydrase (CA) enzymes, falters. The CA enzymes are responsible for balancing the consumption of O2 with the production of carbon dioxide (CO2). Asynchrony here would be problematic. This in turn would stress mitochondria and evoke a whole host of potentially negative reactions. Among them, I think alpha-ketoglutaric dehydrogenase downregulation is a part, but I have no evidence of this yet.
That is what I have thus far. As I continue to put pieces together, I will publish subsequent posts. For now, though, I think the key takeaway is that when replenishing thiamine, we have to be cognizant of the other macro and micronutrients involved in the production of energy, except in acute, immediately life-threatening cases, proceed thoughtfully. What that means clinically, and to those individuals for whom even the slightest amount of thiamine sets off deleterious reactions in the rate, rhythm, or pressure of the heart, I do not yet know. Perhaps others will chime in with theories.
We Need Your Help
More people than ever are reading Hormones Matter, a testament to the need for independent voices in health and medicine. We are not funded and accept limited advertising. Unlike many health sites, we don’t force you to purchase a subscription. We believe health information should be open to all. If you read Hormones Matter, and like it, please help support it. Contribute now.
Yes, I would like to support Hormones Matter.
Feature image by Gerd Altmann from Pixabay.
This article was published originally of February 15, 2021.














Hello! According to my urinaty acid test: high pyruvate, norm lactate, norm citrate, low cis-aconitate, low isocitrate, low alpha-ketoglutarate, normal-high succinate, high fumarate, norm malate; also no deficiency in b6, biotin, inositol, carnitine, b12 according to other urinary acid test parameters (not name it); also possible deficiency in manganese and intermittent copper\zinc\iron\manganese imbalance according to many hair mineral analyses (few years) and blood tests.
Supplementing with b-vitamins, and many minerals make me feel better (no beri-beri now, but no phisical endurance and fatigue very-very quickly), but also over-stimulated state (not always, but frequent), manganese and chrome especially over-stimulate me (but deficient!); can tolerate very-very high dosage of b1, b2, b12 and magnesium – it all make me feel better and my mind is work now; but cant tolerate potassium; and possible sodium-potassium-water imbalance accorrding to many symptoms (dizziness, tachycardia, brain fog, fatigue).
Any help please?!
I would look at sulphur matabolism as well as thiamine has it in it’s structure
Does thiamin “remove “or reduce calcium (plaque )build up in arteries?or is that the job of d3?
Nope, it s the job of vit K2(mk4 form)
I think you might want to add some research on the combined effects of aluminium and fluoride into the mix. I’ve been following the trail of breadcrumbs from covid-19 right through hypothyroidism, vagus nerve impairment, heavy metal poisoning, fluoride and aluminium poisoning right through to disrupted calcium homeostasis, disregulated by ace2, nAchR, Na+ K+ ATP-ase on the entry side, mitochondrial dysfunction on the other side, sarcoplasmatic calcium pump on the third side, and autophagy right in the middle. Aluminium + fluoride affects all of these systems very specifically, unlike heavy metal poisoning which has a much broader palette of disruptions. Aluminiumfluoride (ALF4) mimics phosphorylation and disrupts signalling.
300 mg twice a day is too much too early. You’re experiencing a paradox reaction. Start low and increase slowly. Some folks start at 10 mgs.
There are numerous anecdotes on this site that will offer some insight.
Search this site for the term “paradox”.
Hi Dr. Marrs,
I just started thiamine (2-300mg B1 Hcl) after years of undiagnosed dysautonomic symptoms. I’ve had dizziness, palpitations, air hunger and chest tightness for years which has always been attributed to GERD and bloating. Since starting the thiamine I’ve noticed in increase in these chest symptoms. Do you think this is related to your theories on the cardiac changes? Is this something you have seen resolve with adequate co-nutrient and mineral intake? Perhaps this a part of the paradox that many experience (?)
Thanks for all the work you do! Looking forward to any developments.
300 mg twice a day is too much too early. You’re experiencing a paradox reaction. Start low and increase slowly. Some folks start at 10 mgs.
There are numerous anecdotes on this site that will offer some insight.
Search this site for the term “paradox”.
“With longstanding thiamine deficiency and metabolic dysfunction, the fuel preference of the heart adapts and switches from a primarily fatty acid derived form of oxidation (60-70% fatty acid preference over glucose) to one that is entirely dependent upon large amounts glucose via anaerobic fermentation. The shift back would be difficult, especially if thiamine is given quickly and in large amounts, or possibly any amount, absent the other nutrient co-factors…”
Dr. Marrs, In the event that one is indeed having a difficult time shifting back, is it at least reasonable to assume that the shift back will, in fact, occur? Do your findings indicate what timeframe would characterize this period of transition? Or is there a risk that the cardiac remodeling/looming heart failure outpaces this recalibration?
Jonathan, I wish I knew the answers to these questions.
My gut tells me that the body will heal. It is extremely adaptable. Consider the fuel shift initially was in an effort sustain life. Consider also, that the initial fuel shift was a long term process, not something that occurred overnight. With that, I cannot help but to believe that given the right substrates that it can shift back. How long that will take or how complete it will be is not clear in general, but particularly on the individual level. We each bring to the table a life time of exposures, stressors and survival adaptations.
Of course, all of this is very complicated and I am still working to untangle this particular aspect. I will have another post on it next week and likely several others in the weeks to come as I try to put the pieces together.
Hi again Dr. Marrs,
Thank you for you your response and also for your work on this issue. Interestingly, I had been struggling with some sudden and unprecedented debilitating symptoms without a noticeable cardiovascular component (extreme fatigue, mild psychosis, dizziness, anxiety, POTS, etc.) The “condition” has evolved such that some of those symptoms have receded but I now have a chronically high heart rate, low blood pressure, and frequent (and alarming) SOB/chest-tightness.
Months ago when I read your book, I dismissed the idea that I had wet beriberi and promptly forgot about it despite now matching that exact profile. It’s occurring to me after reading this post that these new symptoms may not be aspects of the initial condition so much as symptoms of the thiamine supplementation I began to treat those conditions. This might explain the shift in symptoms I’ve experienced. Thiamine treated many of the initial problems while introducing a new host of referring problems.
I ascent to the idea that electrolytes and especially calcium seem to be playing a central role in my heart symptoms and my kidneys also seem intimately tied up in the whole thing. I am eagerly awaiting your upcoming posts.
For now, I have two additional questions:
-Related to your idea that issues of gas exchange seem to be involved…have you given thought to the role that chronic usage of masks (covid) might play for these individuals. It could be irrelevant, but I seem to notice a rather pronounced exacerbation of symptoms after wearing my mask for an extended period.
-Fellow powerlifter here (at least, before this illness). What role do you think a structured exercise program could play in “rehabilitating” one’s metabolic physiology to readapt to oxidative phosphorylation? As I understand it, exercise programs can be geared to facilitate an athletes improved metabolic efficiency. Couldn’t the same process that helps make healthy cells more efficient contribute to making maladapted cells healthy? If this were true, then perhaps a structured exercise program incorporating elements of progressive overload and proper recovery could be helpful in accelerating the recovery of the refeeding sufferers you describe.
Regarding the mask issue, I really do not know. I suspect for some individuals the masks can be problematic but I have not done the research on this yet.
Regarding exercise – I absolutely believe exercise is key, however, and this is a big however, it has to be programmed carefully and thoughtfully. Folks have tendency to go all out, then relapse, get worse, and have to begin the recovery process all over again. You might find this case interesting, including some of the embedded hyperlinks on the role of exercise in mitochondrial health. https://www.hormonesmatter.com/narcolepsy-basal-ganglia-mitochondrial-fitness-kickboxing/
Some of her initial symptoms and those with the initial period of thiamine repletion definitely affected her her heart, though the bulk of her issues were brain related. Now, years later, her heart function remains stable, but should she miss her workouts, her brain function lags significantly. With the lockdowns, she did not have access to a gym and trainers and subsequently lost a lot coordination, gait, balance etc. It has been a month, I think, since began training again and much of it is coming back, but she still has a ways to go.
You might also consider my experience with Covid interesting relative to working out. Though I did not have thiamine deficiency in advance and was supplementing throughout, the symptoms very much paralleled a functional thiamine deficiency. https://www.hormonesmatter.com/covid-notes-calcium-tsunami-cardiomyocytes/
The effects it had on my heart and my ability to workout were significant, but I knew that I had to keep moving in order to recover. I had to scale way back though and it took a few months to build back up.
Chandler,
From a mineral balancing perspective, malnutrition slams us into a slow metabolic state, as a basic stress response. In HTMA lab results, Ca will deposit on hair tissue in high amounts (loss from bone), telling us that the body’s stress response is putting us into a state of hibernation in order to survive. What you/we are effectively trying to do is pull someone out of malnutrition and out of a state of hibernation/ very slow metabolic state to a faster metabolic state. We have to move them slowly out of this hibernating state, right? Cuz their body put them into hibernation for survival purposes. To do this we consider supplements that will support their current oxidation rate and modulate their stress/vitality response (usually a good multi, but sometimes they need a lot of sedative minerals and micronutrients Ca, Mg, Zn etc), and they will generally need a good deal of Ca (which helps the body from being overstimulated as well). Adrenal and Thyroid glandular support will certainly have to be entertained as well. As their bodies are gently repleted with nutrition they will also respond better to the nutrition being given….
Wouldn’t Nutritional Ketosis drive this recovery back toward normal physiological function as a matter of necessity?
Chandler, you wrote above: “While glucose is typically contraindicated in the thiamine repletion, as it is often a causative factor in the shifted metabolism in the first place, it is possible that in some instances it may be warranted.”
INDEED. If one has been avoiding carbs, especially anything with actual sugar in it, then one’s glycogen levels may already be tanked. The adverse reactions people are having may be because of the SUGAR IS EVIL that pervades the internet these days.
Yes, EXCESSIVE sugar intake isn’t good, but when you go to the hospital, they don’t hook you up to an agave drip, they use glucose.
Here’s a case study of a woman with anorexia who did improved on thiamine, but after stopping and restarting it, her body temperature didn’t normalize until they “abruptly increased’ the SUGAR in her diet.
https://www.metabolismjournal.com/article/0026-0495(83)90063-X/fulltext
Thanks for your work!
Any updates to Jonathan? I’m experiencing something very similar after starting high dose thiamine.
Hi Dr Marrs,
When I saw “Calcium Management” in the title and realized that your article was about calcium regulation in the heart, I immediately thought of what I had learnt about vitamins D3 and K2 in the last year or so (I first started looking into health matters in mid 2019 after deciding to try and find out what had happened to my elderly mother after she was hit rather badly by a virus in early 2019 from which she has never fully recovered). As I understand it, D3 is necessary for absorption of calcium from food, while K2 ensures that excess calcium is removed from soft tissues and put into bones and teeth, thus together you could say they regulate the level of calcium in soft tissue, including the heart. I notice in your “calcium tsunami” article you say your symptoms started to abate after adding vitamins D and K2.
I’ve often thought there was a curious symmetry between thiamine on the one hand and vitamin D on the other, in that a given condition may be thought by many to be due to vitamin D deficiency while others ascribe it to thiamine deficiency, with each vitamin having its supporters and little thought being given by each camp to the other. An example is Alzheimer’s, which I have often seen connected to thiamine deficiency. But then there is this HM article: “Decreasing the Risk of Alzheimer’s with Vitamin D”. As you will see this article talks about vitamin D ensuring the constancy of calcium. It also mentions magnesium being a partner of vitamin D, as it is of thiamine. Another example would be diabetes, which is said to be associated with thiamine deficiency, while vitamin D is said to improve insulin sensitivity. Yet another example is COVID, which many believe is amenable to vitamin D supplementation, while Dr Lonsdale advocates thiamine and magnesium.
In view of this, I have often wondered if there is a direct connection between the two. Being in the thiamine camp, I wondered if a vitamin D deficiency somehow impedes the normal functioning of thiamine in its role of mitochondrial energy production, thus having an effect similar to an actual thiamine deficiency. From your article, I am wondering if calcium provides the link: vitamin D3/K2 deficiency leads to excess calcium and this has deleterious effects on thiamine as you describe.
Exactly how this might work to bring about the adverse results of attempted thiamine supplementation you describe is beyond my ken, but I wonder if there could be a role for vitamin D3/K2 supplementation in improving the results of attempted thiamine replenishment in affected individuals.
Interesting that you should mention K2. We had a case of a child who had a number of issues related to thiamine deficiency but that could not tolerate thiamine until K2 was on board. Her mom, through research and a fair degree of trial and error figured this out. I had always thought that was interesting and had meant to find the connection. Per your comment, I took a quick look to see how thiamine and K2 interact and apparently there is a somewhat reciprocal relationship, in bacteria at least, but one would expect in mitochondria too. Interestingly enough, the AKG enzyme complex is a central player. Inasmuch as it appears central in Ca2+ regulation, I imagine this is yet additional evidence that negative reactions to thiamine may be related to deficiencies in other nutrients.
And I don’t doubt that vitamin D is deficient in folks who have these reactions. I also suspect that even though the reaction is suggestive of excess intracellular Ca2+ in the heart and elsewhere, my gut tells me that overall they are likely low in Ca2+ and that the excess we are seeing is part related to poor management of the ERs and SRs but part break down of bone and release of stored calcium, likely as a result of the deficient, D,K2, magnesium, thiamine and other micronutrients. So it is much broader than just the thiamine deficiency.
Here are the articles on K2>AKG>thiamine
https://pubmed.ncbi.nlm.nih.gov/1459959/
https://www.biorxiv.org/content/10.1101/841569v1.full
i guess the reationship´between vitamin D and thiamine is because Vitamin D deficiency reducing B vitamin production in the gut
https://pubmed.ncbi.nlm.nih.gov/27515213/
So i guess before we see thiamine deficiency we should see vitamin D deficiency
Javier,
I am just now seeing these comments and yes, this makes sense. I think we forget that the gut produces a notable amount of B vitamins but is easily disrupted by all sorts of variables. So even with supplementation, if we do not fix the gut, we remain, perhaps not deficient, but certainly incapable of maintaining appropriate concentrations of B vitamins. The vitamin D and by association, K2 are involved, is not surprising. That these then influence Ca2+ regulation makes sense.
i guess the reationship´between vitamin D and thiamine is because Vitamin D deficiency reducing B vitamin production in the gut
https://pubmed.ncbi.nlm.nih.gov/27515213/
So i guess before we see thiamine deficiency we should see vitamin D deficiency
Wow. If vitamin D deficiency leads to reduced levels/deficiency of all the B vitamins as the article suggests, that would certainly explain a lot.
How much calcium and magnesium are needed?