

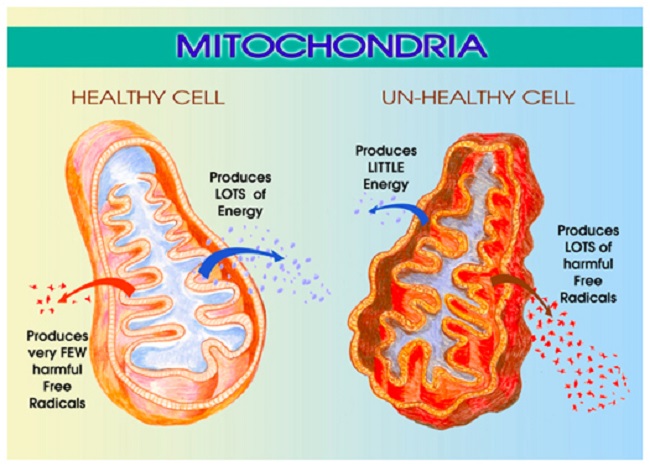
We’ve been learning a lot about mitochondrial dysfunction lately, particularly as it applies to medication and vaccine adverse reactions. Mitochondria are those energy producing powerhouses located within our cells that are critical to every aspect of health. We have over 100,000 trillion mitochondria in our body, each containing 17,000 little assembly lines for making adenosine triphosphate (ATP), the fuel that powers our lives. Mitochondria use ninety percent of the oxygen we breathe, take up 40% percent of the space inside the heart cells and 20% of the space inside the liver cells. Properly functioning mitochondria are critical to human health and survival.
Unfortunately, mitochondria are exquisitely sensitive to their environs and can be damaged easily. Damaged or dysfunctional mitochondria lead to an array of complex and seemingly disparate and untreatable diseases. How mitochondria become damaged is a matter of great interest with research pointing to maternal genetics and epigenetics, environmental exposures, medications and even diet. Mitochondrial dysfunction arguably represents one of the most unrecognized causes of disease in modern medicine and, according to some geneticists, mitochondrial disease represents the next great paradigm in medicine.
The Mitochondrial Cholesterol Transporter
Over the course of my research, I stumbled on a series of papers identifying a cool little transporter channel located on the outer membrane of the mitochondrion called the translocator protein 18kDA or TSPO. For a while, this channel was called the peripheral benzodiazepine receptor because researchers had noticed that the drug diazapam, a benzodiazepine, could bind to a spot on this channel and evoke a reaction. Soon however, researchers learned the function of the TSPO was far more complex than a simple drug binding site and changed the name accordingly.
What is the Mitochondrial Translocator Protein and Why is it Important?
The primary function of the TSPO is to bring cholesterol into the mitochondria. Once inside, another protein, an enzyme called StAR, converts cholesterol into a hormone called pregnenolone and shoots it back out into the cell and beyond. Pregnenolone is the precursor for all steroid hormones and so this TSPO channel is responsible for steroidogenesis in cells, not just in what are considered the typical steroid producing cells like the ovaries, testes, adrenals, but in all cells. TSPO is ubiquitous across mitochondria, meaning steroidogenesis is not limited to the endocrine cells; something many, including myself, have been arguing for decades, but I digress.
In addition to its role in steroidogenesis, the TSPO appears to control many aspects of mitochondrial function. It regulates production of reactive oxygen species (ROS) – those pesky little free radicals that damage mitochondrial DNA. The TSPO also influences outright apoptosis or cell death, and as one might expect, TSPO modulates cellular energy or ATP production. Researchers have found that the functionality of the TSPO channel changes with different disease states and can become dysregulated which then dramatically affects how the mitochondria function. Because of its diverse and seemingly unrelated functions, the TSPO is thought to be part of our host-defense response to disease and injury.
Obesity Impairs Mitochondrial Function via TSPO Downregulation
Here’s where it gets interesting. Once again, we see that diet is critical to mitochondrial functioning via its influence on TSPO activity. In a recent study, Translocator Protein 18 kDa (TSPO) Is Regulated in White and Brown Adipose Tissue by Obesity, researchers demonstrated that a high fat diet downregulates TSPO functioning significantly and the compensatory reaction – obesity – is the survival mechanism.
Study Details. Mice were fed a high fat diet (60% of calories from fat) from 8 weeks of age through 34 weeks of age. Researchers then compared TSPO functioning and other markers of the diet induced obese mice to mice fed a regular diet (13% of calories from fat). They investigated the differences TSPO function and activity in both white fat and brown fat in both groups of mice. Remember white fat stores calories as big fat droplets, while brown fat stores it in smaller droplets and utilizes calories more effectively by burning them for energy. The brown fat is more mitochondrial dense than white fat. So TSPO changes relative to diet and white and brown fat function could be very interesting when understanding obesity.
Results – White Fat. The mice with dietary induced obesity showed a 90% reduction in TSPO mRNA and an 87% reduction in gene expression as measured via a protein called the peroxisome proliferator-activator receptor coactivator (PGC1α). PGC1α is important for mitochondrial biogenesis, making new mitochondria. The researchers also found a 40% reduction in TSPO binding sites, another marker that indicates decreased TSPO gene expression. Interestingly, the shape of the fat cells changed in the diet induced obese mice compared to the regular feed mice. In the obese mice, the white adipocytes were hypertrophic (oversized) and were surrounded by macrophages, the immune cells responsible for ‘eating’ dead or dying cells.
Results – Brown Fat. In the brown fat, the changes in TSPO function between the diet induced obese mice and the regular feed mice were equally dramatic. Visually, the adipocytes were hypertrophic indicating increased fat storage versus energy usage. TSPO gene expression was reduced by 32%, mitochondrial biogenesis was reduced by 31%, while TSPO binding sites decreased by 7%.
A couple more findings. The investigators added a few more conditions to the experiment to determine whether these changes in TSPO could be modified acutely in response to fasting or other stressors. The answer was no. TSPO expression and function were not influenced by acute metabolic changes suggesting a more chronic pattern of metabolic dysfunction.
What This Means: Diet Affects Mitochondrial Function
High fat diet affects mitochondrial function by downregulating a critical receptor – the TSPO channel that brings cholesterol into the mitochondrion. This results in increased cholesterol storage within the fat cells and perhaps decreased conversion from cholesterol to energy (or steroids) inside the mitochondria. The hypertrophic and unhealthy adipocytes then evoke an immune response drawing macrophages and inducing phagocytocis. A dangerous feedback loop ensues.
Though the mechanisms by which a high fat diet induces mitochondrial dysfunction is not clear, one can speculate that a diet high in fat is also low in critical nutrients that are required for proper mitochondrial function and cellular energy production. Deficits in nutrients such as thiamine (vitamin B1) can and do cause severe mitochondrial dysfunction and lead to an array of disease processes from nervous system destabilization to cardiac, GI and reproductive dysfunction. Other nutrients and cofactors are also critical for mitochondrial function. CoQ10, L-Carnitine for example, have been found helpful in mitochondrial induced dysautonomia, and the list goes on.
What’s important to remember is that when the mitochondria are starved of critical nutrients, they don’t function properly and die off. As mitochondria become injured and die, it is possible to see how the hypertrophy and increased cholesterol storage observed in the adipocytes might be a compensatory survival reaction to maintain the requisite demands for mitochondrial cholesterol. The only problem is that as those fat cells become larger and more cholesterol is stored, more mitochondrial dysfunction ensues, reinforcing and continuing the immune response. We get an endless cycle of poorly functioning and dying mitochondria>immune response>larger fat cells with more cholesterol stored>more mitochondrial death>more immune response and so on. Breaking the cycle may be as simple as changing one’s diet and exercising, as both can induce mitochondrial biogenesis.
We Need Your Help
More people than ever are reading Hormones Matter, a testament to the need for independent voices in health and medicine. We are not funded and accept limited advertising. Unlike many health sites, we don’t force you to purchase a subscription. We believe health information should be open to all. If you read Hormones Matter, like it, please help support it. Contribute now.
Yes, I would like to support Hormones Matter.
Photo by Yolanda Djajakesukma on Unsplash.









The main point is that the study was done on rats. Their diets are naturally high carb/low fat. Not applicable to humans I think. Pretty much like that study on cholesterol’s effects where they fed rabbits meat.
I tried to find what was in the HFD; 60% kcal from fat, Cat. no. D12532, Research diets Inc., New Brunswick NJ but was unable to find additional information. Given that many individuals are having success on LCHF and ketogenic diets and studies are emerging to support the benefit of weight loss on high fat fits it would be interesting to know if the diet was made up of trans fats, saturated, MCT or monounsaturated fats as all fats are not created equal and don’t have the same metabolic effects.
Dear miss marrs,I want to thank you for all your hard work.you touch many lives.If it is at all possible I would love to correspond with you.I received 5 lupron shots for breast cancer, because I couldn’t tolerate tamoxefen or arimidex.I could barely walk while on lupron. I had debilitating bone and muscle pain. I cryed all day.chemo put me in menapause. Before lupron I requested an estradiol level which was 22.the doctor should have checked estradiol lab monthly instead of giving me such a powerful drug.He assumed it would go up.I even told him the abd. Ultra sound showed one ovary was shriveled up and they couldn’t find the other one.I need some guidence,I’m 50 years old and with each passing month I have new health issues.please help me. I am suffering and very desperate.is there anything I can do to turn this around?I would be happy if my estradiol was between 50 and 100.
This post I believe does have great information that also may be a bit technical for some to understand. However, being a person with good scientific understanding and also from personal experience I can tell you it is good work you are doing. As I was looking for more information on obesity and mitochondrial disease/dysfunction, being that I have gastroparesis, and a bunch of other issues at times. I asked to be tested for Mito, results are pending. The odd thing is that I am different in that I don’t lose weight during hard times like most GP patients rather my body gains it. So I really do appreciate the science your doing to help people who have Mito dysfunction and related diseases. I am currently getting my Masters in Nutrition and Integrative Health while my undergrad was BS Biology, Minored in Chemistry and Sociology.
Thank you for your comment. It is a technical post – technical subject, but one I think that merits investigation and understanding. For too long we have have equated weight with calories in and out, which is entirely too simple. My body too holds weight and I have been playing with the notion that if I feed my mitochondria, the weight will release. Hasn’t worked yet, but I am a heck of a lot healthier than I was before beginning this. I am glad you are getting tested for mito dysfunction. Remember though, it’s not always about mutations, sometimes there are functional deficits, e.g. nutrient deficiencies that render the mitochondria dysfunctional. Good luck and if you ever want to write about this or your story, send us a note.
I believe that this is a very important post. It is, of course, highly technical and will be over the heads of many who read this. It points to a new way of thinking about common diseases in their relationship to energy metabolism, diet and environmental stress.